The thing about predicting the next mutation in a directed-evolution trajectory is that you have to decide what “right” means. Most antibiotic resistance machine learning predicts phenotype from genome. Given this bacterium’s whole genome, will it be resistant to ciprofloxacin? I wanted to do something different. Given an ordered history of mutations that have already accumulated under selection, what’s the next one going to be? That’s a sequential-decision problem on the evolution side rather than a classification problem on the diagnostic side, and at the start of Day 1 I had not realized how much that framing matters.
Day 1 was the foundation. 28,768 lab-evidence genomes pulled from BV-BRC, annotated with AMRFinderPlus at 98.81% coverage. 17,991 unique resistance protein sequences embedded with ESM2-650M at half precision on an 8GB RTX 4060 Ti. Trajectory data from ALEdb (28,028 transitions across 860 trajectories) plus deep mutational scanning measurements from Pal 2015, Marcusson 2009, and Gullberg 2014 for fluoroquinolone-target proteins. A LightGBM LambdaRank model over candidate mutations hit top-3 = 0.708 overall, which sounded fine. Then I sliced by canonical-AMR mutations specifically — the ones that show up in the CARD database of clinically-relevant resistance — and canonical-AMR top-3 dropped to 0.13–0.21.
The Day-1 test split was a more specific problem. About 86% of the ALE trajectories came from one hypermutator-strain experiment selecting for fluoroquinolone resistance, so the data has wild proportions of passenger mutations riding along with the canonical ones. The held-out e1230 trajectory had n=7 canonical-AMR transitions in it. Anything I did to the canonical-AMR metric on that split was within ±14 percentage points of meaningless.
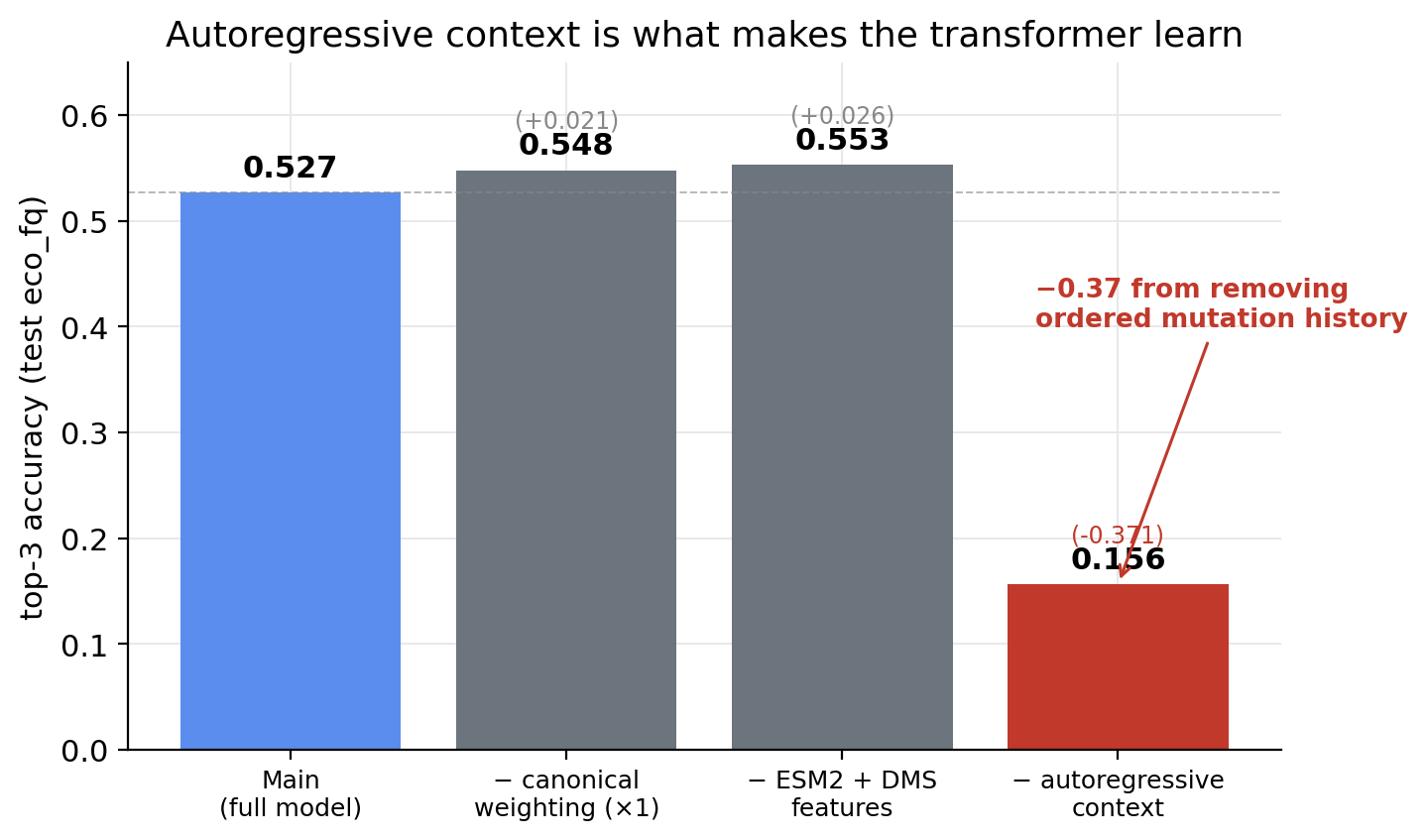
Day 2 I built an autoregressive transformer over the trajectory token sequence. Four-layer causal decoder, d_model=128, taking concatenated learned-mutation-embedding + ESM2-residue-projection + DMS-fitness-scalar inputs, conditioned on drug class and organism. Cross-entropy loss with a 5× weight on canonical-AMR mutations. The headline finding from the day was an ablation. Removing the autoregressive context (predict next mutation from a <START> token only, no history) drops top-3 from 0.527 to 0.156. A +0.37 lift from giving the model ordered mutation history.
That was a clean architectural finding and it survived every form of scrutiny I put it through. The trouble was that the transformer alone at top-3 = 0.527 still underperformed the LightGBM LambdaRank baseline of 0.708 on the same test split. So the transformer wasn’t replacing the LGBM. It was learning something the LGBM wasn’t, and also losing to it on the overall metric.
Day 3 was the realization that the right structure isn’t a single end-to-end model. It’s a reranker.
LightGBM and the transformer fail in opposite ways on full-vocabulary scoring. LGBM gets canonical-AMR top-3 = 0.054 because it ranks all 14,006 mutation tokens additively without considering trajectory context, so it’s dominated by per-class frequency priors. Transformer gets 0.000 because the per-position softmax over 14,006 candidates loses to frequency priors when there’s no candidate filtering at all. So: let the LGBM filter down to the top-10 candidates per transition, score those 10 with the transformer’s autoregressive log-probabilities, and combine them in a second LightGBM that takes both signals plus frequency priors and 14 drug-class one-hots as features.
Canonical-AMR top-3 on the proper rebuilt test split (n=571 transitions across all 14 drug classes, because the Day-2 n=7 split was unusable and I had to rebuild the eval): 0.403.
The per-class breakdown isn’t uniform. Carbapenem and diaminopyrimidine top-3 are 0.67 each. Rifamycin 0.40, glycopeptide and nitrofuran 0.375, fosfomycin and sulfonamide 0.30, macrolide 0.18. That covers eight of the fourteen drug classes. The other six (aminoglycoside, cephalosporin, fluoroquinolone, penicillin, phenicol, tetracycline) show n=0 in the reranker’s per-class output, which is a pipeline bug not a model failure. When the transformer’s vocabulary doesn’t contain a candidate the LGBM proposed, the (mutation, candidate) pair gets dropped from the feature matrix instead of receiving a has_tf=0 indicator. The fix is mechanical, I just didn’t ship it before the project got handed off.
A few other holes worth being honest about. The MIC supervision is on a linear-ramp proxy, because ALEdb provides endpoint MIC measurements but not per-flask values. The apparent Pearson 0.92 between predicted step number and MIC is largely tautological, since the proxy was constructed to be a linear function of step number to begin with. The macrolide canonical-AMR top-1 = 0 on n=11 transitions is biologically suspicious and probably means the mutation tokenization breaks for ribosomal-RNA loci, where most macrolide resistance actually lives. No held-out-organism or held-out-drug-class evaluation, so the 0.403 is on chromosomally-similar data to the training set.
The reranker design — frequency-prior baseline filtered, autoregressive transformer rescored, gradient-boosted combination — is the thing I’d most defend. The data is almost entirely chromosomal point-mutation evolution (1 of 28,028 transitions is plasmid-borne, consistent with ALEdb’s serial-passage origin), so the model is learning chromosomal evolution patterns specifically. That’s a useful narrow regime, not a general-purpose AMR predictor.