The disaster moment came at the end of Day 2.
I had spent two days building a cell-free DNA cancer detection pipeline over the Loyfer atlas. 23 deep-WGBS healthy samples (GSE186458, around 140× coverage), NNLS deconvolution into 40 cell types, in-silico cancer mixtures built from TCGA tumor profiles, a multitask MLP that hit AUROC 0.90 on the in-silico held-out set. The biology checked out. Granulocyte fraction was 29%, which matches Loyfer’s published 30%. Stage-3 sensitivity at 99% specificity was 36.5%, which felt respectable for a first pass. Then I ran the trained model on the only public real-patient cfDNA cancer cohort I could get without dbGaP access: GSE122126, nineteen samples across four cancer types.
AUROC 0.217. Below random. Not “the model didn’t generalize”. Worse than that. The model had learned an inverted decision boundary on real data, and was confidently calling healthy samples cancer and cancer samples healthy.
The kind of result that either kills a project or teaches you something useful, and I had a day left to find out which.
The diagnostic was straightforward once I sat down with it. My model trained on in-silico mixtures generated by linearly combining healthy cfDNA at WGBS coverage with high-coverage WGBS tumor tissue. GSE122126 is Infinium 450K array data, which covers around 10% of the 9,520 atlas blocks the deconvolution operates over. When the classifier sees a sparse 90%-missing input it has never been told what “missing” means, so the imputation defaults to zero and the model interprets that zero as a real measurement of “no signal at this block”. A WGBS-trained model meeting array data is meeting an out-of-distribution input it has no representation for, and out-of-distribution behavior of saturated sigmoids is whatever direction the gradient happened to slope before training ended. In this case, backwards.
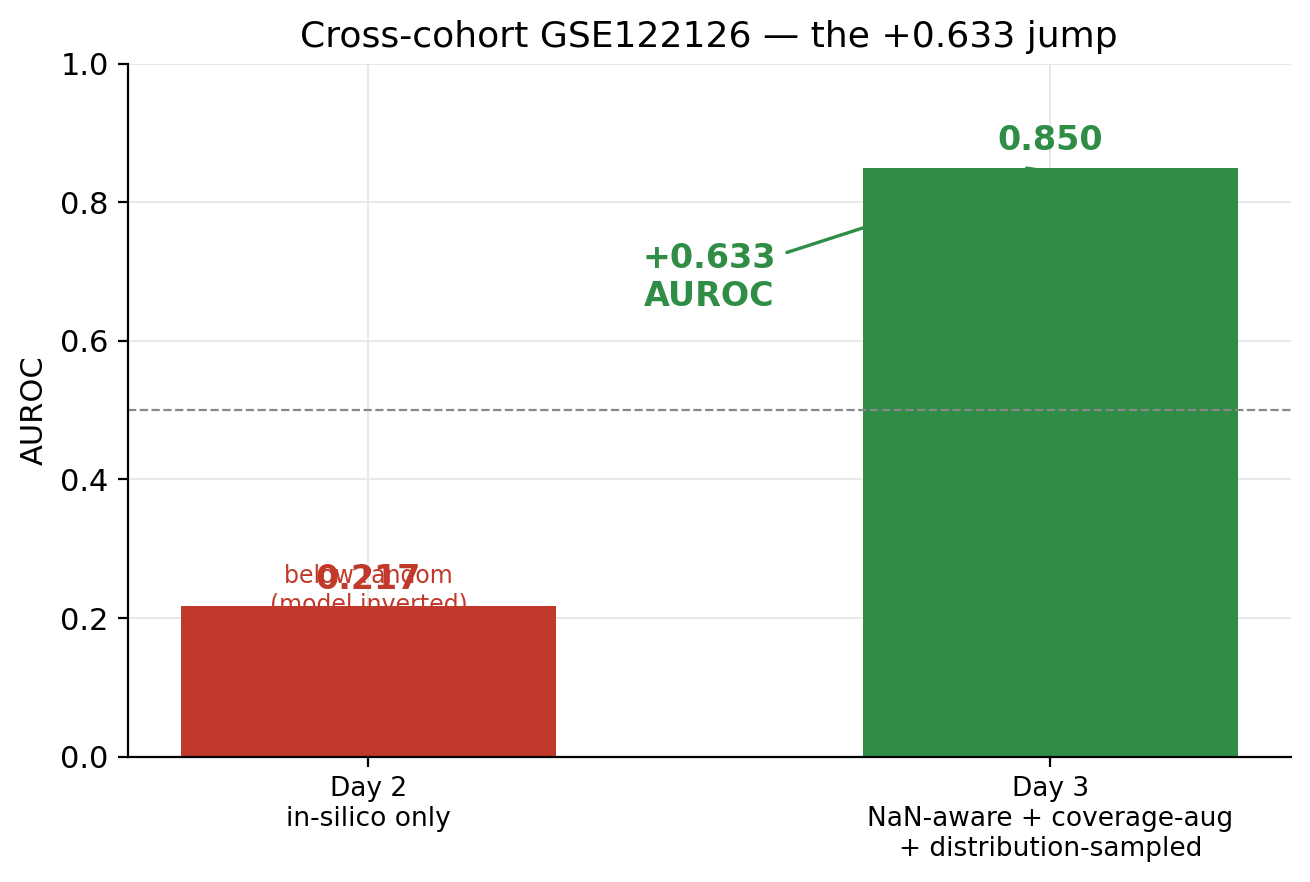
Day 3 was four fixes applied in sequence, with each one exposing the next failure.
The first fix was the training distribution. Instead of mixing one healthy donor with one tumor donor at fixed ratios, I fit per-cancer Gaussian mixture models to TCGA tumor methylation arrays (COAD, LIHC, LUAD, BRCA, with 19 to 30 samples each) and sampled new tumor profiles from those GMMs to mix with healthy cfDNA. The first attempt collapsed completely. TCGA’s 450K arrays cover 8.4% of the atlas blocks, and per-block-mean imputation at the unobserved blocks pulled the sampled tumor profiles back toward healthy means at 91.6% of the input. v2 used the per-block Gaussian where TCGA observed, and fell back to the Loyfer atlas’s tumor-tissue centroid where TCGA didn’t (Colon-Ep for COAD, Liver-Hep for LIHC, and so on). That worked.
The second fix was making the deconvolution model NaN-aware. The input expanded from a 9,520-dim beta-value vector to a 19,040-dim concatenation of imputed-betas and a binary observation mask, so the model could distinguish real measurements from filled-in zeros. Training augmentation: random 10–80% of atlas blocks dropped to NaN per sample, forcing the model to learn coverage-invariant representations of cell-type fractions.
The third fix was coverage augmentation at the classifier head. Each training sample’s deconvolution features were regenerated at four coverage levels (100%, 30%, 15%, 8%) with identical labels across all four. This is the actual trick that made the cross-cohort number work, because GSE122126 lives in the 8–15% atlas-coverage regime and a classifier trained only on full-coverage features had never seen anything resembling sparse input.
The fourth thing isn’t a separate fix exactly. It’s the cleanup that makes the first fix not collapse. I think of it as a fix because I had to discover it the hard way.
Cross-cohort AUROC went to 0.850. By tissue: CRC 0.94 on n=4, BREAST 1.00 on n=3, CUP 0.94 on n=4, LUAD 0.56 on n=4. The LUAD failure is real and worth saying out loud. The four LUNG samples in GSE122126 show identical Lung-Ep-Alveo deconvolution to the four healthy controls. Either those tumors are early-stage low-shedders or atlas Lung-Ep-Alveo and Lung-Ep-Bron don’t separate from baseline at the coverage GSE122126 has. I couldn’t tell from the data which of those is true.
In-silico stage-1 sensitivity at 99% specificity went from 0.04 (Day 2) to 0.48 (Day 3). Distribution-sampled mixtures with TCGA-derived tumor profiles taught the model to recognize smaller tumor signatures than the fixed-donor mixtures did.
After all of that I did the literature check, which is what I should have done at the start.
There’s a paper called M-PACT in Nature Cancer 2026. It does coverage-sparsity augmentation training over 3.2 million in-silico samples, with an ensemble of deep neural networks specialized for low-tumor-fraction and sparse-CpG regimes, for pediatric brain tumor classification from cfDNA. Same paradigm I rediscovered. CUPiD (Nature Communications 2024) does the TCGA-tumor-plus-healthy-cfDNA in-silico mixing across 29 tumor classes with n=143+27 validation. crossNN (Nature Cancer 2025) is literally called “cross-platform DNA methylation classification” and uses masking-rate-based training augmentation. MetDecode (Bioinformatics 2024) is coverage-aware deconvolution. A generalizable HCC framework on bioRxiv from June 2025 does cross-platform WGBS-to-tissue validation and reports the same score-saturation issue I documented as a Day-4 priority.
Which is to say: I spent three days reimplementing 2024–2026 work, and the 0.85 cross-cohort number isn’t a breakthrough. It’s at the credible-claim threshold for in-silico-trained cross-platform-tested cfDNA detection, on a small (n=19) cohort, with one known failure case (LUAD) and uncalibrated scores that saturate near the operating threshold. M-PACT achieved 92% / 88% balanced accuracy on n=79 / n=58 validation cohorts and has St. Jude clinical-cohort access I don’t have.
The things this project actually contributes are reproducibility-flavored, not novel. Everything was built from public sources (GEO, TCGA, the Loyfer atlas) with no dbGaP or EGA cohort access. The failure modes are documented rather than suppressed. The four-mechanism ablation is clean. None of that adds up to a paper, but the work is real and the code reproduces.
Day 4 would be Platt or temperature scaling on the saturated cross-cohort scores, a separate full-coverage tissue classifier to restore the Day-2 tissue-of-origin top-1 of 0.811 (which regressed to 0.247 from multi-task pooling), PAAD harmonization since 37 of 40 TCGA-PAAD files were still downloading when this got handed off, and a real diagnostic on the LUAD failure. Either try the Lung-Ep-Alveo versus Lung-Ep-Bron subtype split, or step away from atlas defaults and pick a lung-cancer-specific marker set entirely.